XPD, à la croisée de la réparation, de la transcription et de la mitose
La protéine XPD est connue pour être l'une des sous-unités de TFIIH, un complexe multiprotéique participant à la réparation de l'ADN et à la transcription des gènes. Dans cette étudie publiée dans la revue Science Advances, les scientifiques montrent que XPD peut aussi jouer un rôle dans la ségrégation des chromosomes lors de la mitose, et ceci indépendamment de sa présence au sein de TFIIH. Cela met en lumière les multiples fonctions jouées par XPD et permet de mieux appréhender l'étiologie de maladies qui lui sont associées.
Le gène XPD (pour Xeroderma Pigmentosum groupe D) code pour une hélicase ATP-dépendante connue pour être l'une des 10 sous-unités de TFIIH, un complexe protéique impliqué dans la réparation de lésions au sein de l'ADN ainsi que dans la transcription des gènes codant pour des protéines. Des mutations au sein de XPD peuvent avoir un effet délétère et être à l'origine de maladies autosomales récessives rares tel que le Xeroderma Pigmentosum (XP, ou maladie des enfants de la lune). Les patients XP développent une photosensibilité, une prédisposition accrue aux cancers cutanés ainsi que des troubles neurologiques (retard mental, déficience cognitive progressive, ataxie). Le XP peut parfois se retrouver associé au syndrome de Cockayne, les patients XP/CS développant en plus des problèmes de croissance.
Durant la réparation de l’ADN, l'activité hélicase de XPD contribue à l'ouverture de la double hélice d'ADN au niveau de lésions provoqués par les ultraviolets, ce qui permet leur élimination. Lors de la transcription, XPD joue essentiellement un rôle structural, permettant à TFIIH de pleinement intervenir en tant que facteur général de transcription pour l'ARN polymérase II.
Mais XPD ne se cantonne pas à sa présence au sein de TFIIH et peut se retrouver associé à d'autres facteurs, sans que l'on connaisse le rôle de tels partenariats. C'est ainsi que les scientifiques se sont intéressés aux fonctions jouées par XPD en dehors du complexe TFIIH. Ils ont observé que la protéine XPD intervient lors de la mitose, en interagissant notamment avec la kinésine Eg5, une protéine motrice essentielle à la formation du fuseau bipolaire. Le partenariat entre XPD et Eg5 est finement orchestré par la phosphorylation de Eg5 via les kinases CDK1 et NEK6 et permet à Eg5 d'être correctement localisé au niveau des fuseaux mitotiques. XPD est lui aussi phosphorylé par NEK6, cette modification post-traductionnelle étant absolument requise pour la pleine fonctionnalité de XPD lors de la mitose mais pas lors de la réparation ou de la transcription.
Ces résultats mettent également en exergue l'implication de défauts mitotiques dans le développement de symptômes observés dans la maladie des enfants de la lune. En effet, les mutations de XPD associées au XP étaient pour l'heure considérées comme affectant essentiellement le processus de réparation des lésions de l'ADN. Or à la lumière des travaux présentés dans cette étude, des défauts de ségrégation des chromosomes pourraient aussi contribuer au développement des phénotypes observés chez les patients. Ces travaux montrent ainsi la polyfonctionnalité de XPD et permettent de mieux appréhender l'étiologie de la maladie des enfants de la lune.
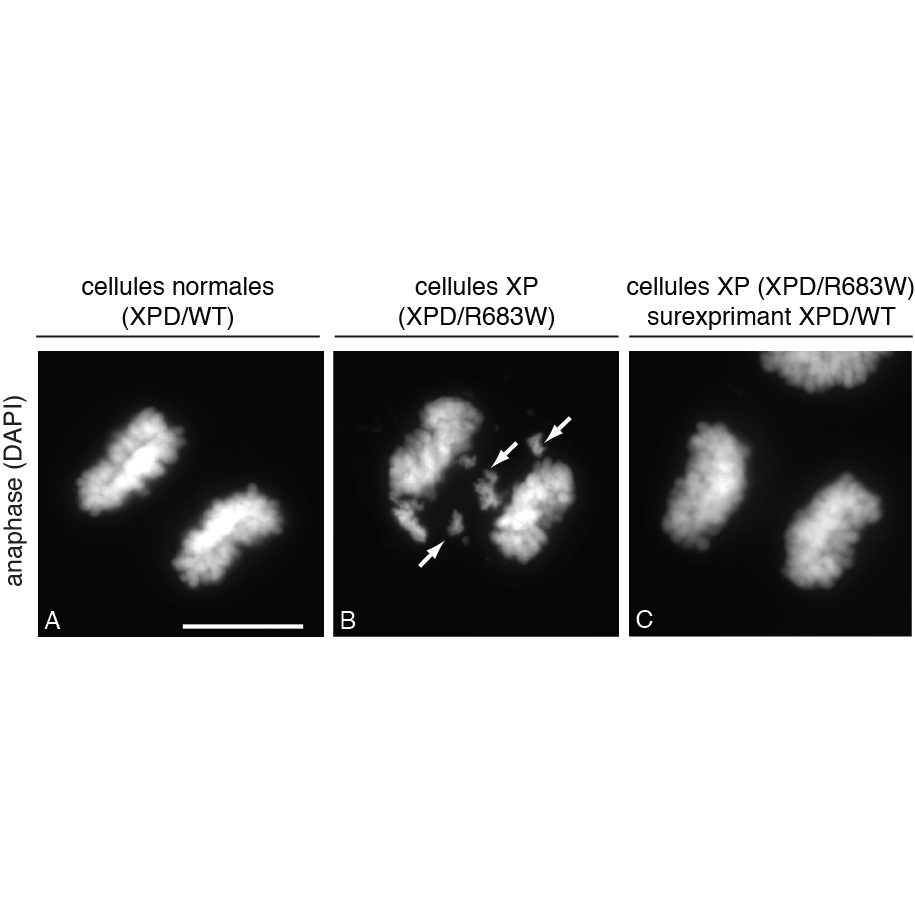
Figure : La ségrégation des chromosomes est affectée lorsque XPD est muté. Marquage au fluorochrome DAPI des chromosomes dans des cellules en anaphase provenant d'un patient sans mutation (A) ou d'un patient atteint de Xeroderma Pigmentosum (XP) et portant la mutation XPD/R683W (B). La surexpression d'une forme sauvage de XPD (XPD/WT) permet de corriger les défauts de ségrégations des chromosomes observées dans les cellules de patients XP (C). Les flèches pointent les défauts d'alignement de chromosomes dans les cellules XP. La barre d'échelle représente 5 μm.
Pour en savoir plus :
Phosphorylation of XPD drives its mitotic role independently of its DNA repair and transcription functions
Emmanuel Compe, Evanthia Pangou, Nicolas Le May, Clémence Elly, Cathy Braun, Ji-Hyun Hwang, Frédéric Coin, Izabela Sumara, Kwang-Wook Choi, Jean-Marc Egly
Science advances DOI: 10.1126/sciadv.abp9457
Contact
laboratoire
Institut de génétique, biologie moléculaire et cellulaire - IGBMC (CNRS/Inserm/Université de Strasbourg)
1 rue Laurent Fries, BP 10142
F - 67404 ILLKIRCH Cedex