Une méthode pour comparer le métabolisme des eucaryotes photosynthétiques
Les réseaux métaboliques à l'échelle du génome permettent de modéliser l'ensemble des transformations chimiques connues au niveau du métabolisme d'un organisme donné. Leur comparaison à large échelle nécessite de s'affranchir des problèmes d'hétérogénéité parmi les génomes disponibles. Dans un article publié dans Genome research, des scientifiques proposent une approche permettant de construire des réseaux de qualité homogène permettant de les comparer dans une perspective évolutive.
Depuis une dizaine d'années, plusieurs cartes du métabolisme des algues ont été reconstruites à partir de données issues de séquençages de génomes. Lors des premières tentatives de comparaison entre ces réseaux métaboliques, les études expliquaient les différences observées entre les cartes métaboliques par l'hétérogénéité des jeux de données issus du séquençage et non par de réelles différences biologiques entre les espèces. La nouvelle approche méthodologique, implémentée dans le logiciel AuCoMe (diffusé sous licence libre), a été développée en utilisant la génomique comparative. Elle permet en s’appuyant sur les connaissances expertes contenues dans les annotations génomiques des espèces les mieux décrites, de déduire les cartes métaboliques des espèces dont les annotations des génomes sont plus rudimentaires – ou incomplètes.
Des gènes manquants ont également pu être identifiés en réalisant des recherches sur les similarités des séquences entre espèces apparentées. Enfin, la méthode intègre aussi une approche pour garantir la fiabilité des prédictions en limitant les biais potentiellement induits par les spécificités d'un génome donné. Cette méthode combine des techniques d'ingénierie des connaissances, de comparaisons de séquences génomiques, et de modélisation de systèmes biologiques pour parvenir à analyser et comparer simultanément le métabolisme de plusieurs espèces eucaryotes, alors que l'état de l'art ne pouvait jusqu'à maintenant que comparer que quelques espèces, et sans pouvoir tenir compte de l'hétérogénéité des génomes.
Une méthodologie qui confirme les prédictions à quelques exceptions près
Cette méthodologie a été appliquée pour étudier une famille d’une quarantaine de génomes d’algues très diverses : plusieurs lignées de microalgues, dont les diatomées et les cryptophytes, ainsi que des macroalgues vertes, rouges et brunes. Elle a permis de d’établir un catalogue unique de cartes métaboliques pour des dizaines d’eucaryotes marins. Les comparaisons entre ces cartes ont montré que les différences entre les métabolismes des différentes espèces étudiées sont pour l'essentiel en accord avec la phylogénie de référence, c'est à dire avec l'état actuel des connaissances sur les relations de parenté entre ces organismes. Il y a cependant certaines différences notables. En particulier, les prédictions des auteurs suggèrent que la microalgue Guillardia theta, considérée comme appartenant au groupe des cryptophytes, dont la position phylogénétique est encore discutée, a un métabolisme qui semble partager des caractéristiques communes à la fois avec les archéplastides (regroupant lignée verte et algues rouges) et les straménopiles (algues brunes et diatomées).
Cette méthode permet d'obtenir rapidement un premier aperçu du métabolisme d'un groupe d'organismes donné, ouvrant ainsi la voie à des études plus approfondies sur certaines spécificités nécessitant de faire l'objet d'études expérimentales complémentaires.
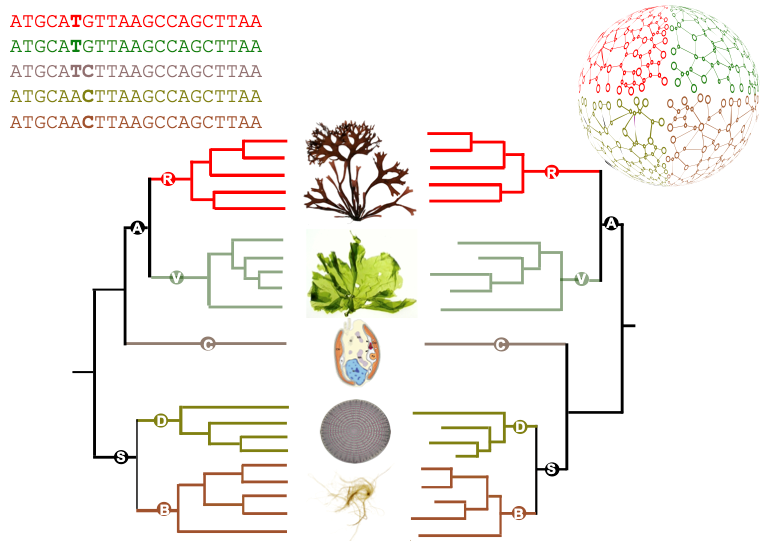
Figure : Comparaison entre une phylogénie de référence générée à partir de séquences (partie gauche) et le dendrogramme métabolique correspondant, issue de la comparaison des réseaux métaboliques (partie droite). Simplifié d'après l'article.
En savoir plus :
Inferring and comparing metabolism across heterogeneous sets of annotated genomes using AuCoMe. Arnaud Belcour, Jeanne Got, Méziane Aite, Ludovic Delage, Jonas Collén, Clémence Frioux, Catherine Leblanc, Simon M. Dittami, Samuel Blanquart, Gabriel V. Markov and Anne Siegel. Genome Research, juin 2023. DOI : https://doi.org/10.1101/gr.277056.122
Contact
Laboratoires
Laboratoire de biologie intégrative des modèles marins - LBI2M (CNRS/Sorbonne Université)
Station Biologique de Roscoff
Place Georges Teissier
29680 Roscoff
Institut de recherche en informatique et systèmes aléatoires - IRISA (CNRS/Université de Rennes)
IRISA, Campus universitaire de Beaulieu
263 Avenue du Général Leclerc - CS 74205
35042 RENNES Cedex - France