Syndrome de Rahman : quand la chromatine se relâche
Le syndrome de Rahman est une maladie génétique rare associée à des troubles du neurodéveloppement et à des traits du visage caractéristiques, pouvant également s’accompagner d’anomalies du squelette ou du cœur. Dans un article publié dans Nature Communications, des scientifiques montrent que la mutation responsable de cette maladie empêche une protéine, l'histone H1.4, de compacter correctement l'ADN. Cette désorganisation pourrait modifier l'expression de nombreux gènes au cours du développement.
H1.4, une histone de liaison qui compacte la chromatine à l’origine du syndrome de Rahman
Le syndrome de Rahman est une maladie génétique rare associée à des troubles du neurodéveloppement et à des traits du visage caractéristiques, avec des manifestations cliniques très variables pouvant associer une apparence de vieillissement prématuré, des anomalies cardiaques ou squelettiques, ou encore des troubles visuels ou comportementaux. Il est causé par des mutations du gène codant l’histone H1.4, l’une des histones de liaison présentes chez l’homme. Cette famille de protéines contribue à organiser la chromatine et influence ainsi la compaction de l’ADN et son accessibilité dans le noyau.
Dans le noyau, l’ADN est enroulé autour de protéines appelées histones dites « de cœur » pour former les nucléosomes, les unités de base de la chromatine. Les nucléosomes sont reliés entre eux par de courts segments d’ADN. Contrairement aux histones de cœur, les histones de liaison comme H1.4 se fixent au niveau des points d’entrée et de sortie de l’ADN sur le nucléosome. H1.4 agit ainsi comme une sorte d’agrafe moléculaire : elle aide à rapprocher ces deux segments d’ADN et stabilise une structure plus compacte de la chromatine. Cette compaction influence l’accessibilité de l’ADN aux machineries moléculaires qui lisent, copient ou réparent l’information génétique.
H1.4 contient une longue région flexible riche en charges positives, qui facilite son interaction avec l’ADN et la compaction de la chromatine. Les mutations associées au syndrome de Rahman modifient cette région et réduisent sa charge positive. Pour cette étude, publiée dans la revue Nature Communications, les scientifiques se sont intéressés à la mutation la plus fréquemment identifiée chez les patients atteints de ce syndrome.
Une chromatine plus ouverte et un ADN plus accessible
Pour comprendre l’impact de cette mutation, les scientifiques ont combiné plusieurs approches complémentaires : des expériences sur des chaînes de nucléosomes reconstituées en laboratoire, des analyses de la chromatine dans des cellules et des simulations numériques.
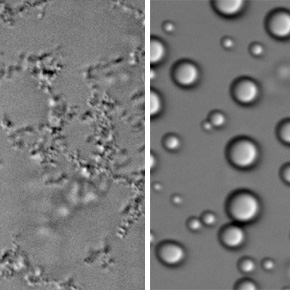
Les résultats montrent que la forme mutée de H1.4 rapproche moins efficacement les segments d’ADN reliant les nucléosomes. La chromatine adopte alors une organisation plus ouverte et plus flexible, où les nucléosomes sont moins régulièrement empilés et où l’ADN devient plus accessible. Les scientifiques ont également observé que cette mutation modifie les propriétés physiques de la chromatine. Alors que la protéine normale favorise la formation de condensats d’aspect fibreux par séparation de phases, la protéine mutée réduit fortement la capacité des chaînes de nucléosomes à former des condensats, qui prennent alors plutôt l’aspect de gouttelettes sphériques. L'ensemble de ces observations montre que la mutation affaiblit la fonction structurante de H1.4.
Vers une meilleure compréhension des maladies liées à la chromatine
Ces travaux établissent un lien direct entre une mutation de H1.4 et une désorganisation de la chromatine susceptible de modifier l'expression de nombreux gènes au cours du développement. Ils apportent ainsi une première explication moléculaire aux anomalies observées chez les patients atteints du syndrome de Rahman.
Au-delà de cette maladie rare, cette étude met en évidence le rôle central des histones de liaison dans l'architecture tridimensionnelle du génome. Elle fournit un cadre pour mieux comprendre d'autres maladies liées à des défauts d'organisation de la chromatine et ouvre de nouvelles perspectives pour l'étude des mécanismes qui contrôlent l'expression des gènes.
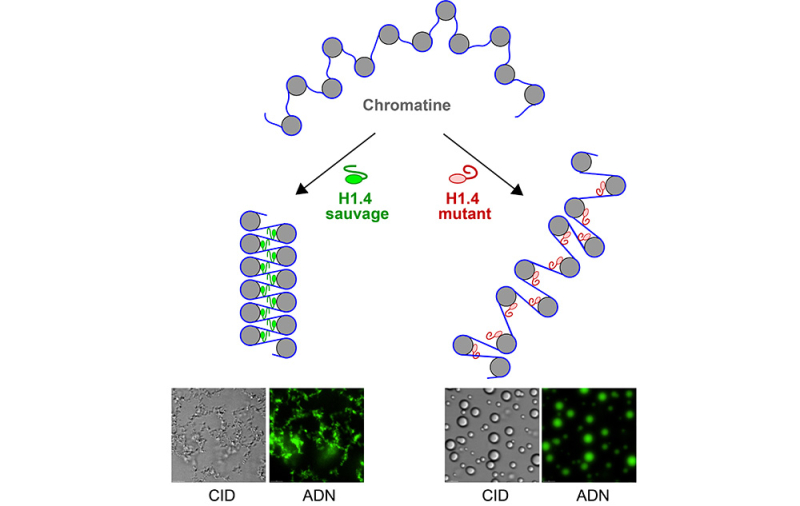
Figure : Schéma comparant l’effet de l’histone H1.4 de type sauvage et de sa forme mutée sur des chaînes de nucléosomes. L’ADN, représenté par des traits bleus, s’enroule autour d’histones de cœur figurées par des cercles gris. La forme sauvage de H1.4 (en vert) favorise la compaction de ces chaînes et, en séparation de phases, la formation de condensats compacts d’aspect fibreux. À l’inverse, la forme mutée de H1.4 associée au syndrome de Rahman (en rouge) affaiblit cette compaction et modifie leur comportement en séparation de phases, conduisant ici à la formation de gouttelettes sphériques. Les images de microscopie montrent les condensats formés par ces chaînes de nucléosomes : le contraste interférentiel différentiel (CID) les rend visibles, tandis qu’un marquage fluorescent confirme la présence d’ADN dans ces condensats.
En savoir plus : Boopathi R, Garcia-Saez I, Turunç S, Lone IN, Kumar A, Abu Alhaija AA, Hayes JJ, Bednar J, Diril MK, Iliev D, Gospodinov A, Le Roy A, Skoufias D, Angelov D, Hamiche A, Kale S, Dimitrov S, Petosa C. A Rahman Syndrome mutation in histone H1.4 disrupts chromatin compaction and phase separation. Nat Commun. 2026 May 22. doi: 10.1038/s41467-026-73046-8. Epub ahead of print. PMID: 42173878.
Contact
Laboratoires
- Institut de biologie structurale - IBS (CEA/CNRS/Université Grenoble Alpes)
71 Avenue des Martyrs
38000 Grenoble - Institut pour l’Avancée des Biosciences (IAB) - (CNRS/ Inserm/ Université Grenoble Alpes)
Site Santé, Allée des Alpes
38700 La Tronche Institut de génétique et de biologie moléculaire et cellulaire - IGBMC (CNRS/Inserm/Université de Strasbourg)
1, rue Laurent Fries
67404 ILLKIRCH CEDEX