scPRINT : le premier modèle d'IA français pour déchiffrer les réseaux génétiques
Chaque cellule est un véritable monde moléculaire. Pour comprendre son fonctionnement, il faut reconstituer le réseau d'interactions entre les gènes qui gouverne son comportement. Dans une étude publiée dans Nature Communications, des scientifiques présentent scPRINT, un modèle d'intelligence artificielle entraîné sur plus de 50 millions de cellules humaines et animales. Son objectif : prédire les réseaux géniques à l'origine des états cellulaires.
Un nouveau modèle fondateur pour la biologie cellulaire
Avec l'explosion des données de séquençage d'ARN à cellule unique (scRNA-seq), les scientifiques disposent d'une cartographie fine de l'expression des gènes dans des millions de cellules. Mais exploiter cette masse d'information pour en extraire les mécanismes sous-jacents reste un défi. scPRINT (pour "single-cell PRe-trained Inference of Networks with Transformers ") répond à ce défi en apprenant, sans supervision, les réseaux d'interactions entre gènes propres à chaque type cellulaire.
Qu’est-ce que scPRINT ?
scPRINT est un modèle de type "transformer", similaire aux modèles de langages comme ChatGPT et LeChat. Bien que plus petit, il est pré-entraîné pour la première fois avec diverses tâches spécifiques à la biologie cellulaire, comme le débruitage des données d'expression génétique, la classification de types cellulaires ainsi que la compression d'information cellulaire. Cela lui permet de représenter finement l'état d'une cellule et d'en inférer les relations géniques. Il intègre aussi de nombreuses connaissances intégrées à sa structure, aussi appelés biais inductifs, comme la position des gènes dans le génome, la structure de leurs produits protéiques, ou encore la statistique intrinsèque des données.
Un modèle explicable et prêt à l'emploi
Contrairement à d'autres grands modèles de cellules, scPRINT fournit des réseaux de régulation génique. Ces prédictions sont obtenues de façon non supervisée, c’est-à-dire que le modèle n’a jamais eu d’information sur les réseaux génétiques durant son apprentissage. Ces réseaux montrent cependant une concordance avec les données expérimentales issues de la littérature, mais révèlent aussi de potentiels nouveaux liens, propres à certains contextes biologiques ou pathologiques. Ces réseaux rendent aussi le modèle plus explicable, permettant de comprendre les liens de causalité que le modèle utilise pour réaliser ses différentes prédictions.
scPRINT est utilisable en "zero-shot" c’est-à-dire qu’il peut annoter des types cellulaires, corriger les variations dues aux différentes techniques de séquençage (effets de batch) ou améliorer les données d'expression sans nécessiter de ré-entraînement. Dans des benchmarks publics, il rivalise avec les meilleurs outils spécialisés, tout en fournissant une sortie plus riche et interprétable.
Une application à l’hyperplasie bénigne de la prostate
Les auteurs ont appliqué scPRINT à un atlas de tissus prostatiques précancéreux. Le modèle identifie une sous-population rare de lymphocytes B exprimant des marqueurs du microenvironnement tumoral. Chez les fibroblastes, il détecte un réseau centré sur PAGE4, impliqué dans la réponse au stress oxydatif, l’inflammation et la fibrose via des échanges d’ions métalliques. Ces résultats illustrent la puissance du modèle pour explorer les changements subtils dans les tissus en dérégulation.
Une ressource ouverte pour la communauté
scPRINT est aussi le premier grand modèle de cellules disponible en open source sur github: https://github.com/cantinilab/scPRINT, avec les poids du modèle, les données ainsi que la méthode complète de pré-entraînement. scPRINT peut être affiné sur de nouvelles tâches par la communauté et être adapté à d’autres espèces ou conditions. Utilisable avec des ressources matérielles standards, il ouvre la voie à une nouvelle génération de modèles d'intelligence artificielle plus explicables et performants pour l’analyse des données single-cell.

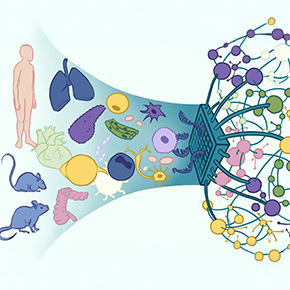
Figure : scPRINT peut recevoir des données d’expression génique à l’échelle cellulaire provenant de différentes espèces et prédire ex nihilo une version nettoyée de ces données, ainsi que des informations sur le type cellulaire, la maladie, l’âge, le séquenceur utilisé, et bien plus encore. Il peut également prédire des réseaux de gènes propres à chaque cellule, qui reflètent des interactions connues dans la littérature scientifique.
À droite, un exemple avec 2,5 millions de cellules prélevées aléatoirement dans la base de données cellule × gène, visualisées, annotées et analysées par scPRINT, qui a également inféré leurs réseaux de gènes.
Référence : scPRINT: pre-training on 50 million cells allows robust gene network predictions. Kalfon, J., Samaran, J., Peyré, G. et al.
Nature Communications, 16 avril 2025, DOI : https://doi.org/10.1038/s41467-025-58699-1
Contact
Laboratoire
Département Biologie du Développement et Cellules Souches (CNRS/Institut Pasteur)
INSTITUT PASTEUR
25-28 rue du Docteur Roux
75724 PARIS CEDEX 15 - France
Département de mathématiques et applications de l'ENS - DMA (CNRS/ENS PSL)
ENS - PSL
45 Rue d'Ulm
75230 PARIS CEDEX 05 - France