
L’intelligence artificielle et les modèles de langages protéiques pour déchiffrer les génomes viraux
Les bactériophages, virus des bactéries, jouent un rôle clé dans les écosystèmes microbiens et potentiellement dans la lutte contre les bactéries résistantes aux antibiotiques. Une étude publiée dans Nucleic Acids Research Genomics and Bioinformatics présente une approche innovante utilisant l’IA et des modèles de langage protéiques pour générer une empreinte digitale de 25 000 génomes viraux. Cette méthode permet d’organiser hiérarchiquement les virus, de révéler leurs liens évolutifs et fonctionnels, facilitant la découverte et la comparaison des virus.
Les bactériophages, acteurs clés du monde microbien
Les bactériophages, virus des bactéries, façonnent les communautés bactériennes et offrent des perspectives prometteuses pour le traitement des infections multirésistantes aux antibiotiques. La taxonomie des phages et leur classification sur la base de liens évolutifs représentent un défi de taille en raison de leur évolution rapide, de leurs échanges génétiques fréquents et du volume sans cesse croissant de nouveaux génomes provenant d’environnements très divers (e.g., le microbiote intestinal, les sols, les océans, etc.).
HieVi : une approche innovante de génomique comparative basée sur l’Intelligence Artificielle
Dans un article publié dans la revue Nucleic Acids Research Genomics and Bioinformatics, des scientifiques présentent HieVi (Hierarchical Viruses), une méthodologie innovante de génomique comparative des bactériophages basée sur l’utilisation de l’intelligence artificielle (IA), plus précisément des grands modèles de langage appliqués aux protéines. Ces derniers dérivent des modèles de langage humains qui ont donné naissance aux IA génératives connues du grand public telles que Le Chat (Mistral AI) ou ChatGPT (OpenAI). De la même façon que ces IA apprennent les langages humains à partir de gigantesques corpus de textes, un modèle de language protéique est capable d’apprendre le language des séquences protéiques à partir de centaines de millions de séquences.
Ces modèles permettent de classer les protéines dans un espace sémantique pertinent en biologie, c’est-à-dire structural et fonctionnel. En faisant l’hypothèse simplificatrice qu’un bactériophage peut être assimilé à un sac de protéines dont les séquences sont codées dans le génome viral, les scientifiques ont utilisé dans leur étude le modèle de language protéique ESM-2 afin de générer de façon non-supervisée une « empreinte digitale » pour chaque bactériophage d’une base de données contenant environ 25 000 génomes complets de virus.
Ils ont montré que cette empreinte encode des informations fonctionnelles et évolutives qui permettent d'organiser les génomes des phages de manière globalement conforme à la taxonomie existante mais qui réserve également quelques surprises qui ont été analysées. Cette approche facilite la découverte de nouvelles familles de virus et permet de classer un nouveau virus sans avoir recours à des approches fastidieuses de comparaisons de séquences.
HieVi constitue ainsi une étape vers une organisation évolutive et facilement consultable des données génomiques des phages, utile pour affiner la taxonomie existante et explorer le paysage complexe et en constante expansion du monde viral.
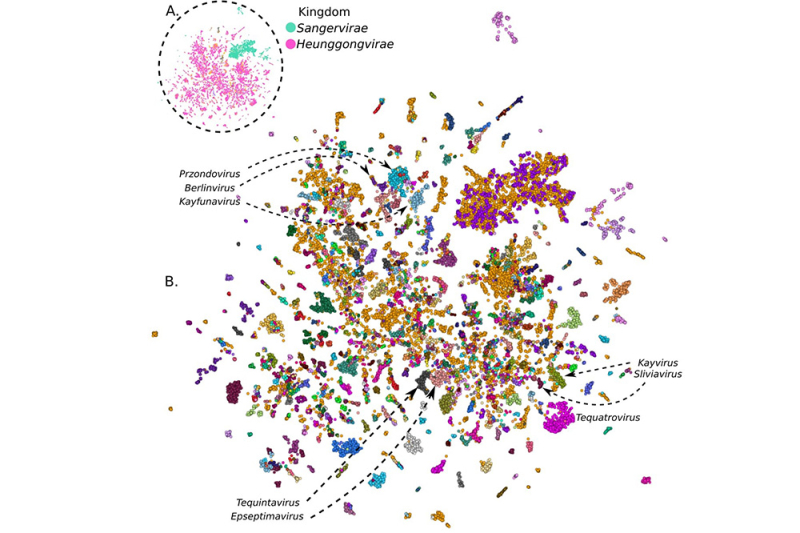
Figure : HieVi Phage Atlas. Projection en deux dimensions de l’espace contenant les représentations vectorielles générées par HieVi pour 24 362 génomes complets de virus procaryotes. Cette projection montre leur regroupement selon des critères taxonomiques et donc des liens évolutifs. Chaque point représente un virus. Panneau A : Vue d’ensemble où les virus sont colorés selon leur royaume (le plus haut rang taxonomique de la classification de l’International Committee on Taxonomy of Viruses). Panneau B : Vue détaillée où les virus sont colorés selon leur genre (le plus bas rang taxonomique avant l’espèce). Les flèches indiquent quelques genres de bactériophages illustrant la cohérence des clusters issus de notre méthode d’analyse.
En savoir plus : Swapnesh PANIGRAHI, Mireille ANSALDI & Nicolas GINET, Phage evolutionary relationships emerge from protein Language Model-based proteome representation - Nucleic Acids Research Genomics and Bioinformatics, https://doi.org/10.1093/nargab/lqaf134
Contact
Laboratoire
Laboratoire de chimie bactérienne - LCB (CNRS/Aix-Marseille Université)
31 Chemin Joseph Aiguier
13009 Marseille