
ProteoCast : prédire l'impact des mutations
Comment distinguer, parmi les centaines de millions de mutations ponctuelles possibles dans un génome, celles qui altèrent réellement la fonction des protéines ? Dans un article publié dans Nature Communications, des scientifiques décrivent ProteoCast, une méthode computationnelle qui parvient à prédire quelles mutations risquent de perturber des fonctions biologiques essentielles. Validée expérimentalement sur le génome de la mouche drosophile, cette approche automatisée ouvre de nouvelles perspectives pour la génomique fonctionnelle et l’identification de variants pathogènes.
ProteoCast : l’évolution comme guide pour prédire l’effet des mutations
Chaque protéine porte en elle l'empreinte de milliards d'années d'évolution. Certaines positions dans sa séquence restent quasiment inchangées d’une espèce à l’autre, signe qu’elles sont essentielles à sa fonction. D’autres tolèrent davantage de variations. C’est sur cette logique évolutive que repose ProteoCast, une nouvelle méthode capable d’anticiper l’impact fonctionnel de mutations génétiques à grande échelle, typiquement les milliers de protéines à l’échelle d’un organisme.
Le principe consiste à comparer, pour chaque protéine, des dizaines de milliers de séquences homologues issues de multiples organismes vivants. À partir de ces alignements de séquences, ProteoCast construit un véritable « paysage mutationnel » : pour chaque position d’une protéine, l’outil estime dans quelle mesure le remplacement d’un acide aminé par un autre risque d’altérer la fonction biologique.
Une classification automatique en trois catégories
Pour rendre ces prédictions exploitables, ProteoCast classe chaque variant en trois catégories : neutre, modéré ou impactant ; grâce à un modèle statistique qui s'adapte automatiquement à la distribution des scores au sein de chaque protéine. La méthode intègre également un indicateur de confiance fondé sur la qualité et la couverture de l'alignement, permettant d'écarter les prédictions peu fiables. Appliquée au protéome complet de la mouche du vinaigre, Drosophila melanogaster, la méthode a analysé environ 22 000 isoformes protéiques pour quelque 300 millions de mutations potentielles. Les résultats montrent que ProteoCast identifie correctement 85 % des mutations létales et impactantes déjà connues comme ayant un effet fonctionnel majeur, alors que seuls 13 à 18 % des variants observés naturellement dans les populations sont classés comme fortement impactants. Cette différence souligne la capacité de la méthode à distinguer efficacement les mutations délétères des variations biologiquement tolérées.
Validation expérimentale par l'édition du génome CRISPR
Pour aller au-delà de la prédiction, les scientifiques ont guidé des expériences d'édition du génome par CRISPR chez la mouche. Cinq mutations ponctuelles ont été introduites dans le gène Naprt, impliqué dans la biosynthèse d'un coenzyme essentiel (NAD). Les trois variants prédits comme les plus impactants (G497P, E147V et K532G) se sont révélés létaux à l'état homozygote, tandis que les deux variants prédits comme neutres (I618P et A262F) ont produit des mouches viables à l’état homozygote. Ces résultats, obtenus en aveugle par rapport aux prédictions, illustrent la capacité de ProteoCast à hiérarchiser des cibles de modification génomique avec une précision remarquable.
Explorer les régions désordonnées des protéines
Une autre force de ProteoCast est de repérer, au sein des régions protéiques dépourvues de structure 3D stable (dites « intrinsèquement désordonnées »), des segments fonctionnels : sites de modification post-traductionnelle (comme la phosphorylation ou l'ubiquitination) et motifs linéaires courts impliqués dans des interactions protéine-protéine. Sur l'ensemble du protéome de la mouche, 63 % des sites de modification répertoriés se trouvent dans des segments à sensibilité mutationnelle élevée selon ProteoCast. Ces résultats suggèrent que les contraintes évolutives détectées par la méthode trahissent l'existence de fonctions biologiques encore non caractérisées.
Une ressource publique, applicable à tout organisme
ProteoCast est entièrement automatisé et s'applique, sans entraînement préalable, à n'importe quel organisme disposant d'une banque de séquences protéiques. Les prédictions complètes pour la mouche sont consultables en ligne sur https://proteocast.ijm.fr/drosophiladb/. Testé également sur des variants humains de la base ClinVar, ProteoCast identifie correctement 77 % des variants pathogènes connus avec une spécificité de 87 %, performances comparables aux meilleures méthodes actuelles. Cette approche évolutive, interprétable et sans apprentissage supervisé, constitue un complément précieux aux outils de séquençage à haut débit pour la génomique fonctionnelle et l'identification de cibles thérapeutiques.
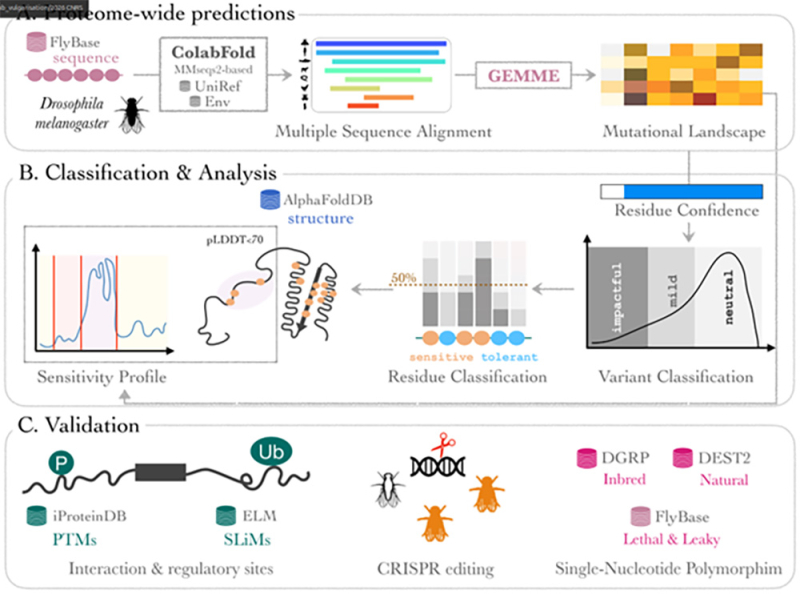
Figure : Représentation schématique du processus d'analyse ProteoCast. A La chaine d'analyse commence par la génération d'un Alignement multiple de séquences (MSA) pour chaque séquence de protéoforme de Drosophila melanogaster à l'aide de ColabFold reposant sur MMseqs2, interrogeant UniRef et des bases de données de séquences environnementales. GEMME produit ensuite une matrice L×20 représentant le paysage mutationnel complet en simple substitution de la protéoforme, où L désigne le nombre de résidus d'acides aminés. B ProteoCast écarte les prédictions peu fiables (confiance par résidu, fondée sur l'évaluation du MSA d'entrée), puis classe les variants restants comme impactants, modérés ou neutres (classification des variants) en ajustant un modèle de mélange gaussien à la distribution des scores bruts. Il détermine ensuite si chaque résidu est sensible ou tolérant aux mutations en fonction de sa proportion de variants impactants (classification des résidus). ProteoCast projette par ailleurs la sensibilité mutationnelle par résidu (score brut moyen) sur les modèles tridimensionnels issus d'AlphaFold DB. Il segmente le profil de sensibilité mutationnelle afin d'identifier de potentiels sites de liaison et de régulation au sein des régions non structurées. C Validation de ProteoCast par des expériences d'édition CRISPR et par confrontation à des sites connus de modifications post-traductionnelles (PTM) et de motifs linéaires courts (SLiMs), ainsi qu'à des polymorphismes nucléotidiques, qu'ils soient observés dans des populations consanguines ou sauvages, ou associés à des phénotypes délétères tels que la létalité développementale ou une perte de fonction partielle.
En savoir plus : Abakarova M, Freiberger MI, Liehrmann A, Rera M, Laine E. Proteome-wide prediction of the functional impact of missense variants with ProteoCast. Nat Commun. 2026 Apr 27;17(1):3813. doi: 10.1038/s41467-026-72140-1. PMID: 42045226; PMCID: PMC13121731.
Contact
Laboratoires
- Laboratoire biologie fonctionnelle et adaptative - BFA (CNRS/Université Paris Cité)
4 rue Marie-Andrée Lagroua Weill-Hallé
75205 Paris Cedex 13 - Biologie Computationnelle, Quantitative et Synthétique - CQSB (CNRS/Sorbonne Université)
4 place Jussieu
75005 Paris Cedex 05