Un gène qui protège contre le vieillissement mais favorise le cancer
La recombinaison homologue joue un rôle essentiel dans la réparation et la stabilité du génome. L’acteur principal de ce mécanisme est la protéine RAD51. A l’aide d’un modèle murin où l'activité de recombinaison homologue de RAD51 a pu sélectivement inhibée: les scientifiques montrent, dans une étude publiée dans EMBO Journal, que contrairement à ce qui était admis, l'altération in vivo de la recombinaison homologue médiée par RAD51, ne favorise pas le développement de cancers, mais au contraire le prévient. Cependant, elle induit un vieillissement prématuré.
L'instabilité génétique est une caractéristique aussi bien du cancer que du vieillissement. La recombinaison homologue est un mécanisme cellulaire qui joue un rôle essentiel dans la réparation et le maintien de la stabilité du génome. RAD51, qui se lie sur l'ADN endommagé avec l’aide de protéines médiatrices telles que BRCA2, joue un rôle primordial à l'étape centrale de la recombinaison homologue. C’est en effet RAD51 qui recherche les homologies de séquence avec un séquence d'ADN intacte qui sera recopiée, servant donc de matrice pour réparer l’ADN endommagé.
En raison de son importance dans le maintien de la stabilité du génome, la recombinaison homologue est généralement considérée comme un mécanisme de suppression des tumeurs. En accord avec cette théorie, de nombreux gènes de recombinaison homologue (notamment BRCA1 et BRCA2) sont mutés dans les tumeurs, en particulier dans les cancers héréditaires du sein et de l'ovaire. De façon surprenante, malgré le rôle central que joue RAD51 dans la recombinaison homologue, son inactivation dans les tumeurs n'est cependant pas observée, ce qui constitue le "paradoxe RAD51". Une hypothèse s’appuie sur le fait que les protéines accessoires, telles que BRCA1/2, favorisent la fixation de RAD51 sur l'ADN endommagé ; les mutations affectant ces protéines entraînent donc l'absence de RAD51 sur l'ADN endommagé, qui devient alors accessible à d'autres processus de réparation mutagènes, augmentant de ce fait l'instabilité génétique. Par conséquent, l'hypothèse mentionnée ci-dessus soulève la question suivante : la formation d’un cancer résulte-t-il uniquement de l'ablation de la recombinaison homologue, ou la stimulation concomitante de voies mutagènes est-elle nécessaire ?
SMRad51, un outil expérimental unique
Les modèles de souris sont des outils utiles pour étudier le développement de cancer ainsi que le vieillissement in vivo. Cependant, RAD51 est un gène essentiel et son inactivation entraîne une létalité embryonnaire précoce. Dans ce travail, les scientifiques ont tiré parti d'une forme dominante négative de RAD51 (SMRad51) qui se lie à l’ADN avec RAD51 et inhibe l'activité de recombinaison homologue du gène RAD51 endogène. Cependant, comme SMRAD51 se fixe à l'ADN endommagé, elle empêche toujours la stimulation des voies alternatives de réparation mutagène. SMRad51 représente donc un outil expérimental unique permettant de séparer l'activité de recombinaison homologue de la protection contre les mécanismes mutagènes de réparation.
Les scientifiques ont conçu un modèle de souris avec une expression conditionnelle ubiquitaire (c’est-à-dire dans toutes les cellules de l’organisme) de SMRad51. Ce gène ne peut s’exprimer dans ce modèle qu’en présence de la Doxycycline (Dox). Cette stratégie a permis de surmonter le problème de la létalité embryonnaire puisque la recombinaison homologue peut être inactivée après la naissance par l'induction de l'expression de SMRad51 médiée par la Dox, ajoutée dans la nourriture.
SMRad51 révèle un effet contre le développement du cancer du sein
Ces souris représentent donc les premiers spécimens pour analyser les altérations du fonctionnement de RAD51 in vivo. La suppression de la recombinaison homologue par l'expression de SMRad51 entraîne un stress réplicatif, une inflammation systémique, l'épuisement des cellules souches progénitrices, un vieillissement prématuré et une réduction de la durée de vie, mais, étonnamment, pas la formation de tumeurs. Pour mieux comprendre le rôle joué par SMRAD51 dans le cancer in vivo, le modèle de souris a été croisé avec celui de prédisposition au cancer du sein PyMT. L'expression de SMRad51 diminue à la fois la fréquence et la taille des tumeurs mammaires, révélant un effet contre le développement du cancer du sein.
L’importance de RAD51 dans l'équilibre entre le cancer et le vieillissement
Bien que les dogmes en vigueur considèrent que la déficience en recombinaison homologue soit une cause de cancer, ce travail montre que la réduction spécifique de la recombinaison homologue médiée par RAD51, sans stimulation concomitante des voies de réparation alternatives mutagènes, conduit principalement au vieillissement et non à l'oncogenèse, et même prévient le développement cancéreux. Ces données mettent en lumière la séparation et la concurrence potentielle plutôt que la coopération entre vieillissement et oncogenèse in vivo. Ces travaux soulignent l'importance de l'activité de RAD51 pour l'homéostasie des cellules souches, la prévention du vieillissement et, plus généralement, pour l'équilibre entre le cancer et le vieillissement.
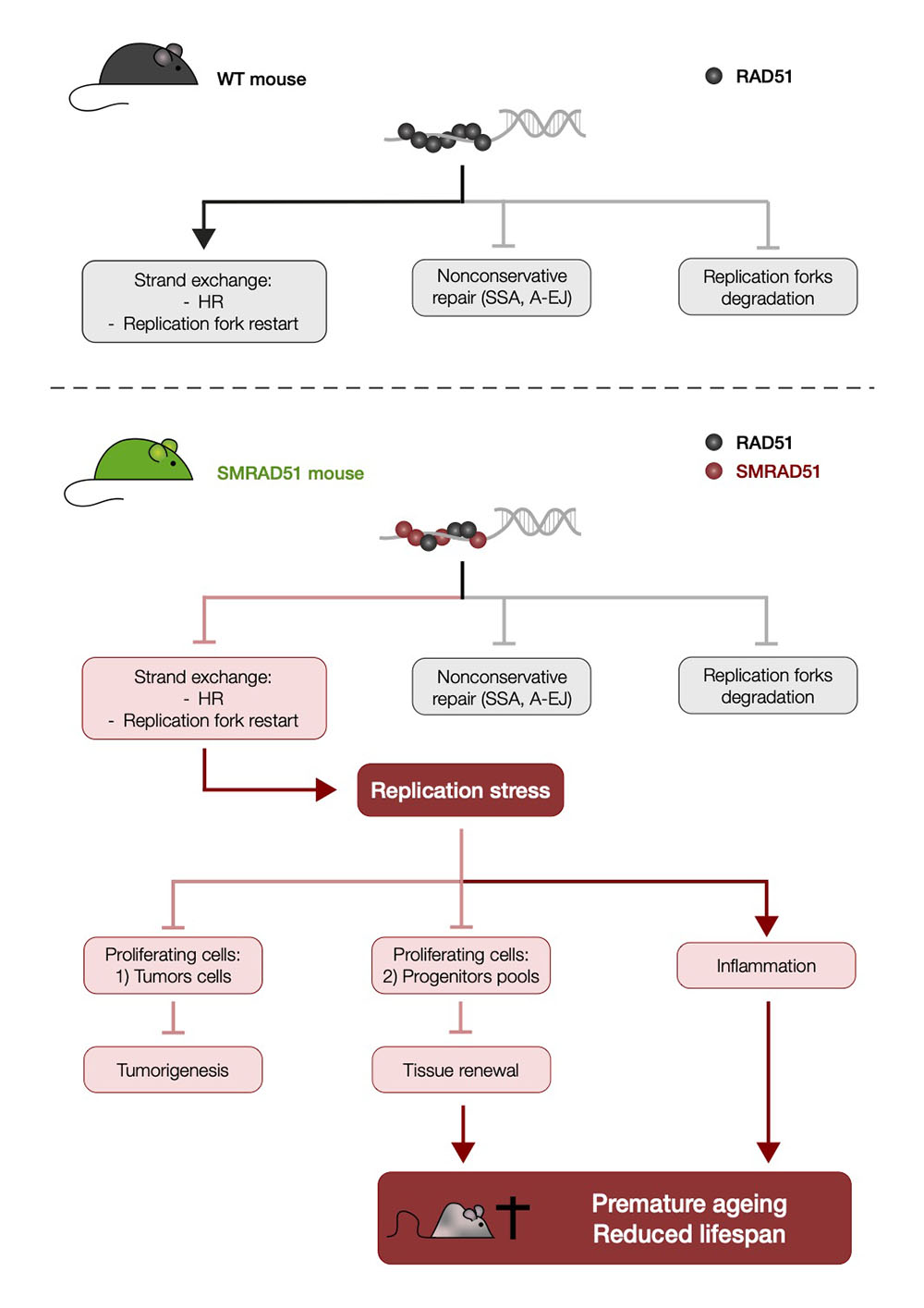
Figure : Impact du RAD51 in vivo. La protéine RAD51 sauvage (panneau supérieur) se lie à l'ADN endommagé, empêchant la réparation mutagène (SSA et A-EJ), protégeant les fourches de réplication arrêtées de la dégradation et favorisant l'échange de brins homologues. L'échange de brins conduit à la recombinaison homologue (RH) et favorise le redémarrage des fourches de réplication arrêtées. SMRAD51 (panneau inférieur) se lie à l'ADN endommagé, empêchant la réparation mutagène (SSA et A-EJ) et protégeant les fourches de réplication arrêtées de la dégradation. Cependant, SMRAD51 inhibe l'échange de brins, altérant ainsi à la fois la RH et le redémarrage des fourches de réplication arrêtées, ce qui entraîne un stress de réplication. Le stress de réplication affecte les cellules en prolifération, donc les cellules tumorales ainsi que les cellules souches progénitrices, entraînant à la fois une répression du développant tumoral et l'épuisement des pools de cellules souches progénitrices. La diminution des réserves de cellules souches progénitrices empêche le renouvellement des tissus, entraînant un vieillissement prématuré. Les déficiences simultanées et combinées de différents tissus réduisent la durée de vie. En outre, l'inflammation systémique (qui peut résulter du stress de réplication) peut également amplifier le vieillissement prématuré et la réduction de la durée de vie.
En savoir plus :
Gabriel Matos-Rodrigues Vilma Barroca, Ali-Akbar Muhammad, Elodie Dardillac, Awatef Allouch, Stephane Koundrioukoff, Daniel Lewandowski, Emmanuelle Despras, Josée Guirouilh-Barbat, Lucien Frappart, Patricia Kannouche, Pauline Dupaigne, Eric Le Cam, Jean-Luc Perfettini, Paul-Henri Romeo, Michelle Debatisse, Maria Jasin, Gabriel Livera, Emmanuelle Martini, Bernard S. Lopez. In vivo reduction of RAD51-mediated homologous recombination triggers aging but impairs oncogenesis (2023). EMBO Journal (in press). doi.org/10.15252/embj.2022110844
Contact
Laboratoires
Institut Cochin (CNRS/Université Paris Cité/Inserm)
24 rue du Faubourg St Jacques
75014, Paris
Institut de biologie François Jacob (CEA/Université Paris Cité/Université Paris-Saclay)
CEA-FAR, Bât 05 p.B0011
18 route du panorama
92265 Fontenay aux Roses